
This is the subject of my teachings, and I go on to describe
all the possible causes of the stars' movement across the Universe.
Of these hypotheses, only one must be the correct explanation,
but what animates the bodies in the sky is still beyond the grasp
of those who move at an honest pace through the path to Truth.
Titus Lucretius Carus, On the Nature of Things, V 529-533
Welcome to my personal blog! What follows is a list of the categories of this website. Click on the title to see the articles it contains. A list of categories is available on the left of this page too (if you are browsing on a PC).
- Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
- Medicine (POTS, Lyme disease, etc)
- Sciences (articles about various disciplines: mathematics, life sciences, mechanics, etc)
- Paolo Maccallini (a collection of autobiographical notes, poems, short stories, and drawings)
A collection of most of my writings on mechanics, mathematics, and biomedicine is available on my Academia page. A collection of my drawings can be found on my Instagram.
Below you find a list of all the articles on this website, ordered from the most recent one to the very first article, published in 2016. Some blog posts are only in Italian, but you can translate them into your language of choice by using the bar on the left.
When I was 20 I developed an unknown condition that drastically reduced my cognitive and physical functioning. As a consequence, I spent most of these last 23 years without being able to concentrate and leaving home. I improve a couple of months per year, on average, and during these improvements, I study my disease, searching for a cure.
As for the quote from the poem On the Nature of Things by Lucretius, an early example of scientific divulgation, the original hexameters in Latin are the following ones:
... id doceo, plurisque sequor disponere causas,
motibus astrorum quae possint esse per omne;
e quibus una tamen sit et hic quoque causa necessest,
quae vegeat motum signis; sed quae sit earum
praecipere haudquaquam est pedetemptim progredientis.
Titus Lucretius Carus, De Rerum Natura, V 529-533
The reader may have recognized some similarities with ideas promoted by natural philosophers born after Galileo had started the scientific revolution, about sixteen centuries apart from the end of Lucretius’ life. Besides the obvious “Hypotheses non fingo” by which Newton admitted that even though his mathematical model described so well the movement of planets, he had no idea of the cause of gravity; besides that, we can also mention Chamberlin’s “Method of Multiple Working Hypotheses” (R) that seems to follow the footprints of these few lines from the fifth book of De Rerum Natura. Verses in which almost a presage of the convergence of a Baconian series can perhaps be acknowledged; a prophecy that has traveled through twenty centuries to teach us what we have painfully learned again during this vertiginous hiatus and that we often forget about.
I leave the reader with a poem I found in the first volume of Don Quijote de la Mancha, the book considered by many the ancestor of all the novels produced by the Occidental world during the last four centuries. Novels are a popular and very recent form of literature: for dozens of centuries, humanity has relied on complex verses to share and convey stories. Verses are hard to write (Virgilius spent on average one day on two hexameters of his Aeneis), and they also require some effort to be fully absorbed; for this reason, I believe that novels are a form of simplified and low-quality literature, even though we must acknowledge their role in shaping modern society. But Literature, whether in verses or in prose, always transmits a story, and a story is always a research. This website contains my story.
Busco en la muerte la vida,
salud en la enfermedad,
en la prisón libertad,
en lo cerrado salida
y en el traidor lealtad.
Pero mi suerte, de quien
jamás espero algún bien,
con el cielo ha estatuido
que, pues lo imposible pido,
lo posible aun no me den.
Miguel de Cervantes Saavedra, Don Quijote de la Mancha I, XXXIII
This poem is found inside the novela “El curioso impertinente”, read by the priest, in the venta, in chapter XXXIII. The author is unknown, but it is believed to be Cervantes himself who, like me, knew first-hand what it means to desperately search for a way out of prison.
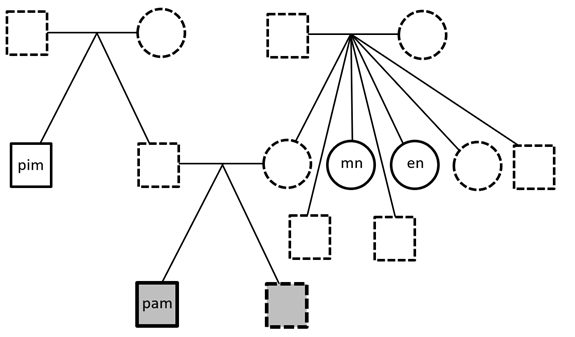
Personal Genomics
Whole Genome Sequencing was performed on the proband (me!) and three healthy family members: one paternal uncle and two maternal aunts. First-degree relatives were not available. Paired-FASTQ files were inputted to a GATK pipeline, according to GATK best practices workflows (R). After the generation of unmapped BAM from paired-FASTQ, a BAM file was obtained for…
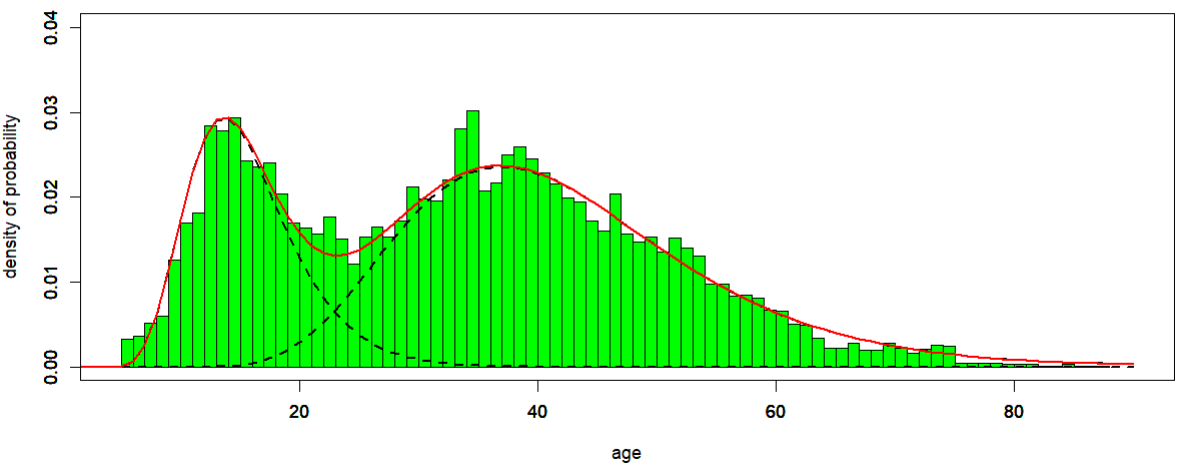
Bimodal distribution of age at diagnosis among ME/CFS patients
The distribution of age at diagnosis for ME/CFS patients follows a bimodal density, i.e. a density with two local maxima (Figure 1), according to the data collected from the Norwegian Patient Register from 2008 to 2012. Data were gathered considering the first G93.3 episode for each patient, and age was calculated as age in 2008…
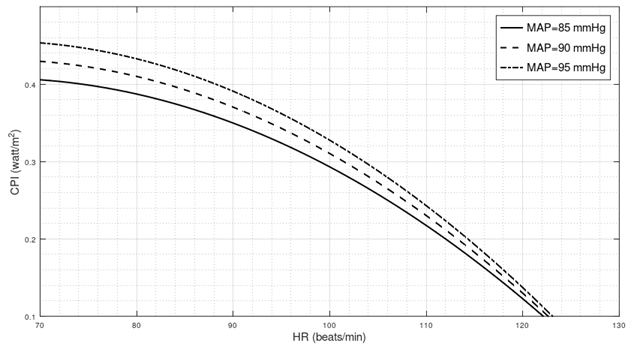
End-tilt cardiac power in Postural Orthostatic Tachycardia Syndrome
Introduction In the following three paragraphs, I present different lines of reasoning suggesting that in a standing position, the left ventricle of POTS patients generates a mechanical power that is lower than normal. Test of significance I considered data from a tilt table testing on 16 healthy subjects and 32 individuals with a previous diagnosis…
Probability Density of Left Ventricular Mechanical Power: derivation from metadata. Application to ME/CFS.
Studies employing tilt table testing are usually accompanied by metadata including means and standard deviations of diastolic blood pressure (DBP), systolic blood pressure (SBP), and cardiac index (CI). But the correlations between these measures are rarely shared and raw data are commonly not publicly available. Therefore, it is impossible to calculate means and standard deviations…
Loading…
Something went wrong. Please refresh the page and/or try again.
Get new content delivered directly to your inbox.
